Lancet (IF=168.9)|关注性别差异,铺路个性化医疗
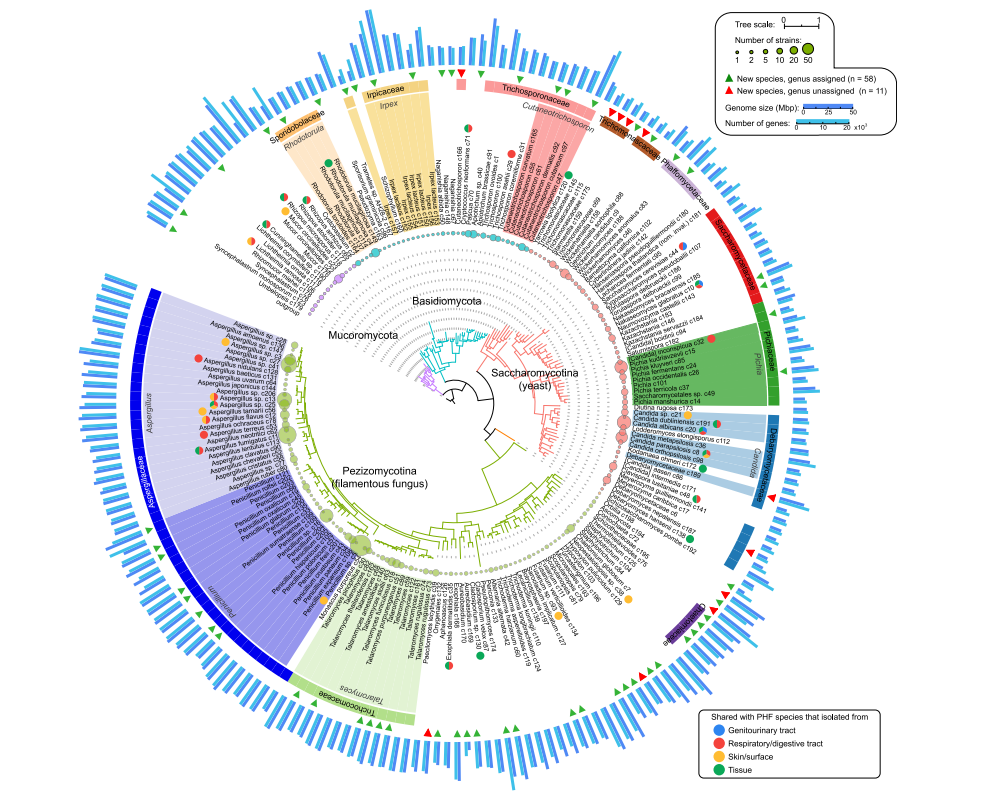
INTRODUCTION•✦研究介绍✦•研究背景Background在过去的二十年里,越来越多的研究开始阐明性别对BMI和肥胖症、情绪障碍和抑郁症、HIV和非传染性疾病等疾病的影响。在未来生物研究中适当考虑性别也逐渐成为常规——尤其是随着分子遗传学、生物医学工程和人工智能为更适合个人需求的治疗开辟了可能性。METHODS•✦研究方法✦•研究指标Outcomes该文参考2021年全球疾病负担研究(GBD),比较了1990年至2021年期间全球七个世界地区10岁以上个人疾病负担的20个主要原因中女性和男性的疾病严重程度(DALY率),DALYs计算为YLDs与YLLs的总和加权,其中YLLs为每10万人的死亡人数乘以死亡年龄的全球标准预期寿命,YLD为疾病或伤害的综合患病率和持续时间。调查范围Seven Regions本调查适用于204个国家和地区从幼儿到95岁以上的20个标准年龄段的女性和男性。根据收入和流行病学概况分为七个区域:中欧、东欧和中亚;高收入国家;拉丁美洲和加勒比;北非和中东;南亚;东南亚、东亚和大洋洲;以及撒哈拉以南非洲。本分析侧重于全球七个地区中每个地区的健康模式,探索不同生命阶段的女性和男性之间疾病负担的差异。见图1.图1. 2021年全球女性和男性DALYs前20个原因的全球排名,年龄标准化(10岁及以上)FINDINGS•✦研究发现✦•1按地区差异分析全球来看女性比男性最常见的疾病是腰痛。我们发现,在南亚女性每10万人比男性多478.5[95%CI 346.3–632.8],性别差异最严重。在所有地区中,女性与男性相比所面临的精神障碍负担更高,比如抑郁症和焦虑症,这种差异在高收入地区的国家最为明显。头痛疾病、其他肌肉骨骼疾病和痴呆症的相对和绝对负担在世界所有地区的女性中都更高。而死亡率(YLL)是导致男性DALY率较高的主要原因,全球七个地区内男性因COVID-19死亡的人数都要比女性多,道路伤害和心脏病紧随其后,特别是在拉丁美洲和加勒比地区以及南亚,男性因道路伤害造成死亡的情况尤为突出。图2. 2021年女性和男性之间DALY率(每10万人口)的全球和区域绝对差异,年龄标准化(10岁及以上)2按年龄层划分道路伤害和精神类疾病的性别差异在生命早期就已开始。在全球范围内在25-49岁女性中,女性在精神、神经和肌肉骨骼疾病方面的差异加剧。除新冠肺炎外,25-49岁人群中观察到的女性超出男性的最大绝对差异是撒哈拉以南非洲的HIV/AIDS。随着年龄的增长,两性之间在腰痛和其他肌肉骨骼疾病的差异持续扩大。另外,在最老年龄组阿尔茨海默氏症和其他类型的痴呆症成为女性的主要疾病负担。相比之下,男性在缺血性心脏病、肺癌和慢性肾病的差异在幼年(10-24岁)时很小,但在一生中持续扩大。而道路伤害一直是个特例,从10岁到49岁,男性在所有地区都承受着更高的DALY比率,只有在老年时有所减少。图3. 2021年,各年龄组女性和男性的全球DALY比率(每10万人口)和95%的UI3时间推移下的变化在1990年至2021年期间,对于这20种疾病来说,女性和男性年龄标准化DALY率之间的全球差异模式随着时间的推移是相对稳定的,但HIV/AIDS出现了巨大的时间差异,从一开始没有明显的性别差异到后来女性患病逐渐低于男性。图4. 1990年至2021年期间,女性和男性之间全球绝对差异的时间模式(每10万人口),年龄标准化(10岁及以上)DISCUSSION•✦研究讨论✦•研究总结1从1990年到2021年,女性和男性之间存在持续的健康差异。我们特别强调了全球女性和男性在各个生命阶段的多样化和不断变化的健康需求,包括女性与发病率相关的条件负担较高,以及男性的新冠肺炎和道路伤害的不成比例负担。2这些发现强调了在卫生系统战略规划中采用生命历程方法的重要性。有效设计和实施卫生系统战略需要了解性和性别之间的复杂相互作用,以及健康的其他社会决定因素。3随着全球人口老龄化,只有通过协调一致的、有性别知情的战略,认识到男性和女性在不同生命阶段面临的独特健康挑战,才能实现人人享有公平和健康未来的进展。参考文献[1] Patwardhan V, Gil GF, Arrieta A, Cagney J, DeGraw E, Herbert ME, Khalil M, Mullany EC, O'Connell EM, Spencer CN, Stein C, Valikhanova A, Gakidou E, Flor LS. Differences across the lifespan between females and males in the top 20 causes of disease burden globally: a systematic analysis of the Global Burden of Disease Study 2021. Lancet Public Health. 2024 May;9(5):e282-e294. doi: 10.1016/S2468-2667(24)00053-7. PMID: 38702093; PMCID: PMC11080072.PROFILEVedavati Patwardhan美国加利福尼亚州圣地亚哥加州大学性别平等与健康中心科学家,跨学科和定量研究人员研究重点是妇女经济赋权、整个生命过程中健康方面的性别差异,以及对低收入和中等收入国家现金转移支付等以女性为中心的政策干预措施的评估。END文案 | 杨思洁排版 | 夏诞审核 | 夏诞发布|姜笑南世界生命科学大会RECRUIT关注我们,获取生命科学学界前沿|促进更多的学术交流与合作业界前沿|促进更快的产品创新与应用政策前沿|促进更好的治理实践与发展
 2024-05-21
2024-05-21